Research Interests
Molecular Dynamics
Structure and Dynamics of Biological Membranes and Membrane proteins
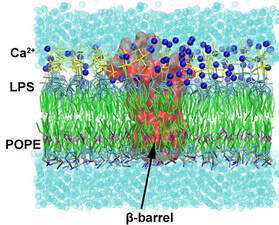
My main interests include the computational study of membrane lipids and transmembrane proteins through Molecular Modeling and Molecular Dynamics simulations. In particular, I use the aforementioned techniques to study the structure and dynamics of lipids, including both standard membrane lipids (phospholipids, sterols, etc) and special lipid classes (e.g. lipopolysaccharides), and transmembrane proteins, including but not limited to, important drug targets such as G-protein coupled receptors (GPCRs), bacterial transmembrane β-barrels, also known as Outer Membrane Proteins (OMPs) and various substrate transporters. I have also created GNOMM, an automated pipeline for the creation of simulation systems of Outer Membranes in Gram-Negative bacteria.
Protein aggregation and amyloid fibril formation
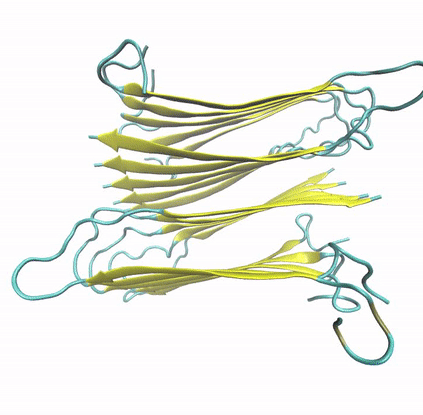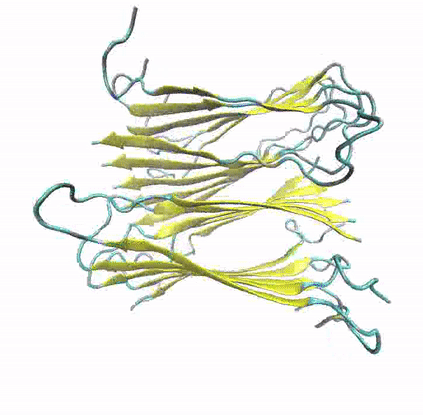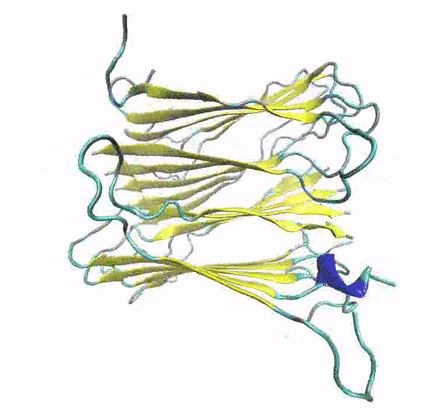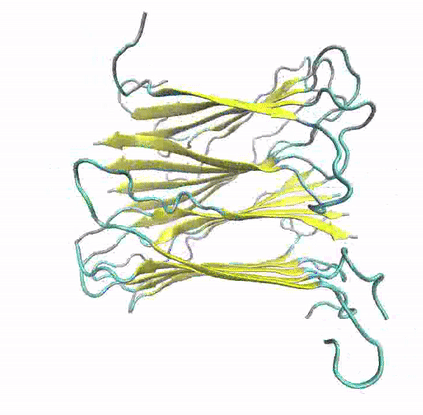
A growing number of proteins and peptides with unrelated functions and no apparent sequence or structure similarities have been associated with the formation of amyloid fibrils via self-aggregation procedures. Highly ordered protein aggregates, the so-called amyloids, are deposited extra- and/or intracellularly in organs or tissues, by adopting a distinct, characteristic architecture. A large number of widespread diseases have been connected with these unrestrained fibril depositions, known as amyloidoses; examples include the Alzheimer’s and Parkinsons’s Diseases, Type 2 Diabetes, Amyotrophic lateral sclerosis (ALS), some types of cancer and many others. However, the structural, dynamic and functional determinants that guide protein aggregation and the manner through which amyloid fibril depositions are implicated in pathological conditions remain controversial.
To explore the structural and dynamic determinants of amyloid aggregation, I have employed Molecular Dynamics simulations on various proteins and peptides, aiming to determine the factors that influence their self-aggregation and the formation of amyloid fibrils. Specifically, I utilize MD simulations to explore the dynamics of self-aggregating peptides during the early nucleation stages of amyloid formation, as well as the structure and stability of amyloid protofibrils.
Biological Networks
Cell-specific signaling of G-protein coupled receptors (GPCRs)
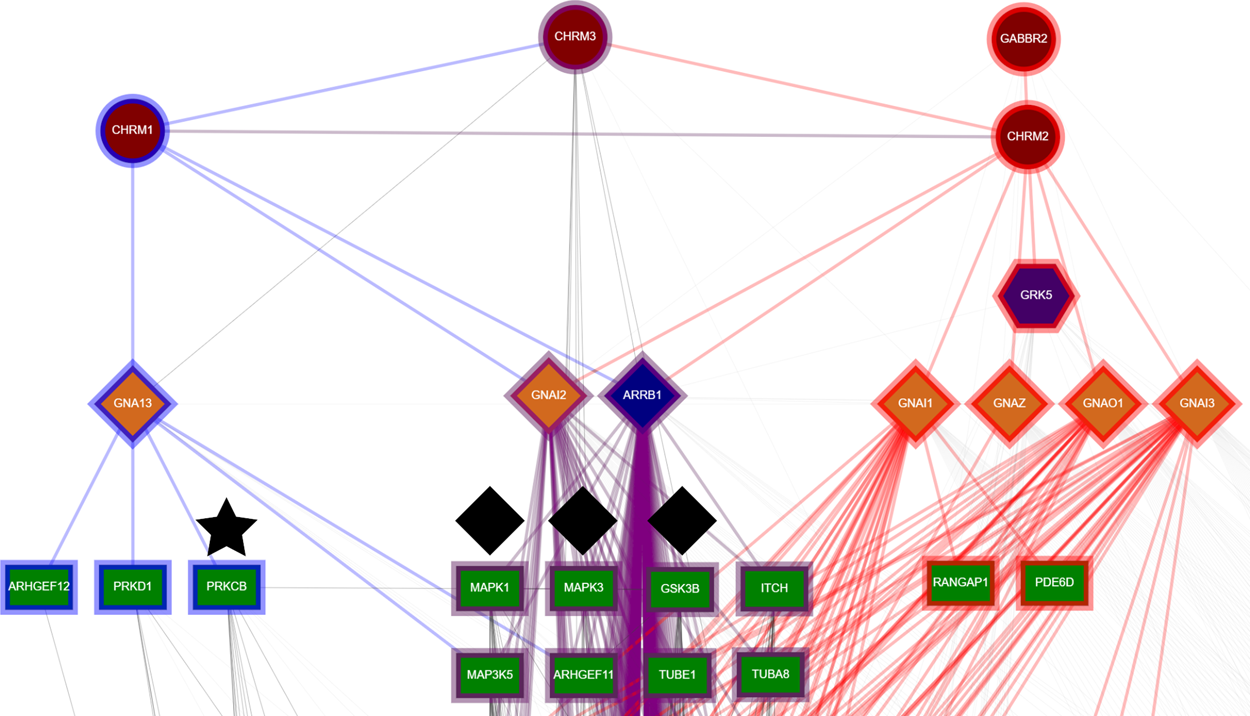
G-protein coupled receptors (GPCRs) are one of the largest and most diverse superfamilies of cell-surface receptors in eukaryotic cells. They recognize a wide array of signaling stimuli, ranging from small molecules and ions to large peptides and steroid hormones to non-biological agents such as smell or light, and regulate the majority of cell responses to these stimuli. As a result, GPCRs have been implicated in several pathological conditions, including neurological syndromes like Parkinson’s disease, schizophrenia, Alzheimer’s disease etc, several metabolic disorders, congestive heart failure, drug addiction, various types of cancer and infection by viruses such as HIV or herpesviruses. As a result, today GPCRs are targets for more then 30% of prescribed pharmaceuticals on the market.
GPCRs signal via a multitude of pathways, mainly through G-proteins and β-arrestins, to regulate effectors responsible for cellular responses. At the same time, a number of other singaling pathways exist, such as direct GPCR-effector interactions and GPCR homo- and hetero-oligomerization. The limited number of transducers results in different GPCRs exerting control on the same pathway, while the availability of signaling proteins in a cell defines the result of GPCR activation. The effect the activation of a GPCR has on a cell is determined by the specific signaling pathways utilized. Since each cell may contain only a subset of all proteins, the pathways available to a GPCR are determined by the available signaling proteins. For this reason, integration of protein interaction data with cell type expression data can be beneficial to study how GPCR function is modified according to the type of cell in question. At the same time, the study of GPCR signaling in specific tissues and cell types combined with biased signaling can help guide drug discovery.
To aid in this endeavor, the Extended Human GPCR Network (hGPCRnet) was designed. hGPCRnet presents a network approach in the study of protein-protein interactions of GPCR signaling pathways, combined with cell expression evidence from over 70 distinct tissues. It allows for the concurrent study of all major GPCR signaling pathways (G-protein, β-arrestin, oligomerization etc) with the additional integration of cell specificity information, thus leading to a more directed view of GPCR functionality in a specific cell type. This combination of network analysis with cell expression evidence enables a more directed approach in studying GPCR signaling and pharmacology.
Protein-protein interactions in the Nuclear Envelope
The Nuclear Envelope is a double-membrane subcellular component, surrounding the nucleus of eukaryotic cells. It acts as a barrier between the cytoplasm and nucleoplasm, controls the transportation of all substances and macromolecules between the two and participates in organizing the nucleus’s interior components. The nuclear envelope’s membranes contain a large number of membrane proteins that participate in numerous protein – protein interactions. These include both proteins that form macromolecular assemblies responsible for substrate transport and nucleus organization, such as the Nuclear Pore Complexes and Nuclear Lamina, and a large number of other membrane proteins that participate in several biological processes. In fact, it has been estimated that the complexity of the nuclear envelope’s proteome and its interactions rivals that of the plasma membrane. However, while other cellular components, such as the plasma membrane or mitochondria have been extensively studied, the proteomics and interactions of the nuclear envelope remain largely unexplored.
My interests involve studying the proteomics and protein-protein interactions of the Nuclear Envelope and its components at a Systems Biology level. Towards this end, I have employed concepts of Network Theory to describe the interactome of the human Nuclear Envelope’s proteins. I have also created NucEnvDB, a publicly available database of Nuclear Envelope proteins and their interactions that offers users a number of computational tools for the analysis and functional enrichment of these interactions.
Sequence analysis and annotation
Detection and annotation of Receptor Tyrosine Kinases (RTKs)
Receptor tyrosine kinases (RTKs) are an important class of transmembrane receptors in Metazoa. They are single-spanning transmembrane proteins, with an extracellular N-terminus containing various domains that recognize peptide ligands and a cytoplasmic C-terminus containg a tyrosine kinase domain. RTKs recognize peptide hormones such as insulin and various growth factors and regulate several cell functions including gene expression, cell growth, apoptosis and programmed cell death. As such, they have been implicated in various diseases, including several types of cancer, neurological syndromes and metabolic deficiencies. Despite their importance inside the cell, RTKs have only been experimentally studied in humans and selected model mammalian species. Conversely, very little is known regarding the existence and function of RTKs in the proteomes of other organisms.

RTK-PRED is an automated method for the detection and annotation of RTKs based on sequence alone. The method utilizes profile Hidden Markov Models (pHMMs) in combination with α-helical transmembrane topology prediction and performs both the detection of putative RTKs and a complete annotation of their topology, as well as a functional annotation of the receptors based on their extracellular domains. RTK-PRED produces high values for sensitivity (100%), specificity (99.70%) and accuracy (99.72%) and is, therefore, a suitable method for the automated detection of putative RTKs in novel, unstudied proteomes. It should be noted that RTK-PRED is, currently, the only publicly available method for the prediction of RTKs from their sequence.
Text mining
Biological text mining and analysis in multiple file types
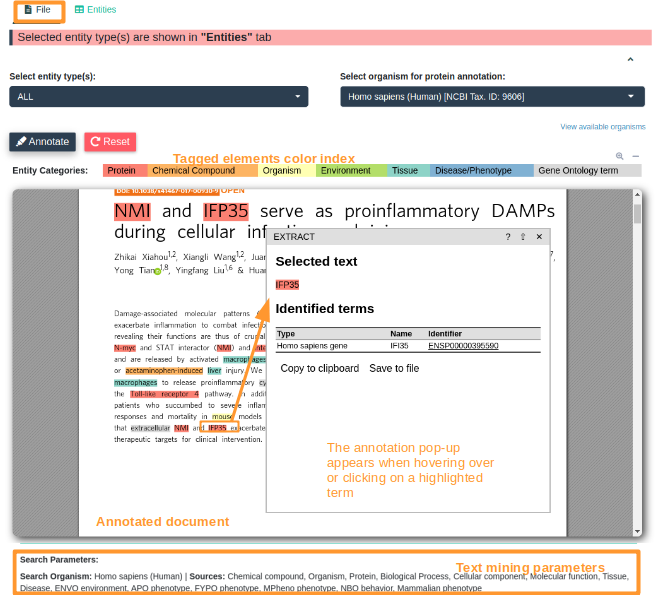
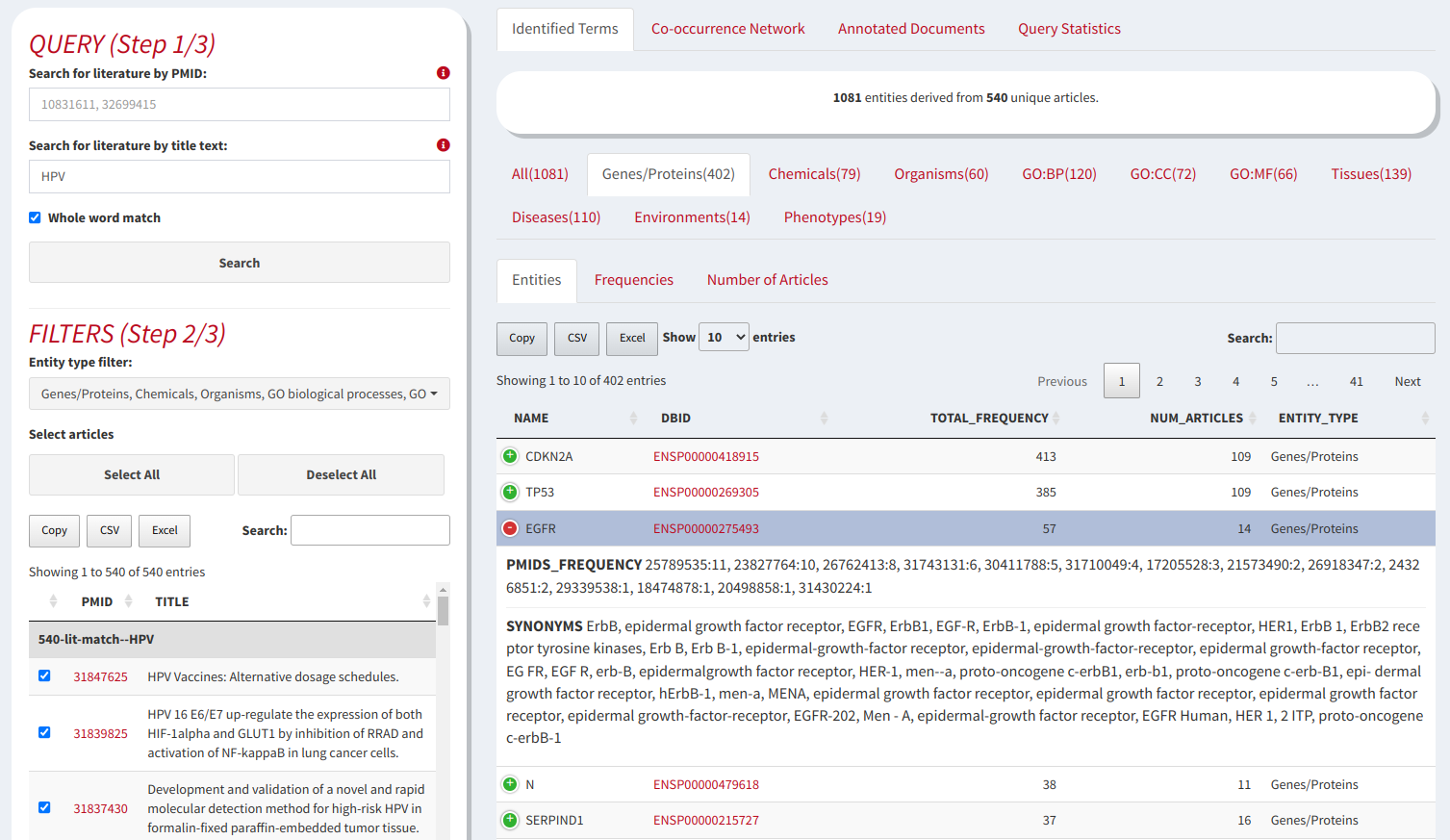
Retrieving all of the necessary information from databases about bioentities mentioned in an article is not a trivial or an easy task. Following the daily literature about a specific biological topic and collecting all the necessary information about the bioentities mentioned in the literature manually is tedious and time consuming. Towards this end we have implemented OnTheFly2.0, a method for the automated extraction and analysis of biological terms from documents. OnTheFly2.0 is a web application mainly designed for non-computer experts which aims to automate data collection and knowledge extraction from biological literature in a user friendly and efficient way. OnTheFly2.0 is able to extract bioentities from individual articles such as HTML, plain text, Microsoft Word, Excel and PDF files. Through an intuitive and easy-to-use graphical interface, the text of a document is extensively parsed for bioentities such as protein and gene names, chemical compounds, organisms, environmental entities and ontology terms. Utilizing high quality data integration platforms, OnTheFly2.0 allows the generation of informative summaries, interaction networks and at-a-glance popup windows containing knowledge related to the bioentities found in documents.
Studying disease-bioentity associations through text mining
Finding, exploring and filtering frequent sentence-based associations between a disease and a biomedical entity co-mentioned in disease-related PubMed literature is a challenge, as the volume of publications increases. Darling is a web application which utilizes Name Entity Recognition to identify human related biomedical terms in PubMed articles mentioned in OMIM, DisGeNET and Human Phenotype Ontology (HPO) disease records and generates an interactive biomedical entity association network. Nodes in this network represent genes, proteins, chemicals, functions, tissues, diseases, environments and phenotypes. Users can search by identifiers, terms/entities or free text and explore the relevant abstracts in an annotated format.